



The majority of people with two MC1R variants had blonde or light brown hair. Several additional red hair colour-associated loci were identified in this study, including variations in the gene encoding the agouti signalling protein (ASIP), an inverse agonist of the MC1R. Over 200 genetic variants were also found to be independently associated with a spectrum of hair colours ranging from black to blond. Many of these associated genes are known to be involved in hair growth and texture rather than in pigmentation, thus highlighting the interplay between melanocytes and keratinocytes in governing hair color [3].
Significant changes in hair pigmentation occur with age, with the darkening of hair colour though childhood, adolescence and young adulthood in response to hormones, including sex steroids and followed by the age-related greying of the hair [4]. This greying, termed senile canities, is usually first observed around the temple and vertex area in men, and around the perimeter of the hairline in women, before spreading over the rest of the scalp [5]. Canities is defined as the progressive loss of natural hair pigmentation over time resulting in a blend of darker normally pigmented hair and lighter hypomelanotic and amelanotic hair, leading to a dilution of hair colour and a gradual whitening perceived as greying [6]. Premature canities can also occur, with an onset as early as during adolescence [7].
Senile canities occurs in both men and women with an incidence in middle-aged adults (45–65 years of age) of 78% in men and 71% in women [8]. The incidence of canities varies depending on ethnic or geographical origin and natural hair colour, with the lowest incidences being reported in populations where dark hair predominates [8]. The average age of onset of senile canities in different ethnic groups ranges from the mid-30s and the late-30s in Caucasians and Asians respectively, to the mid-40s in Africans [4]. Consequently, the age at which greying is defined as premature canities also varies: before the age of 20 in Caucasians, 25 in Asians and 30 in Africans [4].
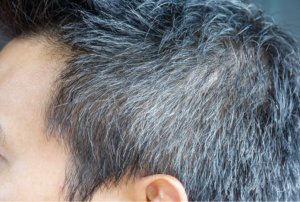

II. Underlying causes of canities
Psychoemotional stress, diseases associated with protein deficiency and malabsorption, and various vitamin (vitamin D3 and vitamin B12) and mineral deficiencies (calcium, copper, and iron or ferritin), have all been associated with an increased likelihood of premature greying [6, 7]. The use of a range of topical agents (dithranol, chrysarobin and prostaglandin F2-alpha analogues) and drugs (chloroquine, mephenesin, interferon-alpha and imatinib) can also trigger premature greying [5]. In addition, environmental factors – exposure to ultraviolet radiation (UVR), pesticides and smoking – influence the onset of canities [6].
Genetic factors play a major role in canities. Premature greying in the absence of any underlying pathology is common and is inherited as an autosomal dominant trait. In addition, numerous pathologies are associated with premature greying, including autoimmune diseases (vitiligo, pernicious anaemia and thyroid disorders), premature ageing syndromes (progeria, Werner syndrome and Rothmund–Thomson syndrome), atopic diathesis and Down syndrome [5, 6, 9].
II. Treatments
Premature canities is a common motivation for seeking consultation with a dermatologist. Reversal of the hypopigmentation associated with some nutritional deficiencies has been reported with an appropriate treatment for the underlying aetiology, such as iron supplementation in patients with anaemia [7]. Numerous treatments have been marketed for canities and can contain a range of compounds (para-aminobenzoic acid, phytic acid, capixyl, calcium pantothenate etc.) but outcomes are often inconsistent and only temporary [6]. The mainstay of treatment is often to camouflage white hair with dyes, with some studies showing that hair colorants protect the hair against photodamage [6, 9]. More promising treatment approaches involve topical antiaging/antioxidant compounds, the recombinant human growth hormone therapy and the use of the α-melanocyte-stimulating hormone (α-MSH) agonist, acetyl hexapeptide-1 (Melitane™) [6, 9]. Future therapies will rely on the identification of target hair follicle genes and proteins, and may also use liposomes to selectively deliver topical treatments to the hair follicle [6].
II. THE BIOLOGY OF HAIR PIGMENTATION
I. Melanocyte development and THE hair follicle pigmentary unit
Follicular melanocytes are derived from melanocyte stem cells which express cell lineage-specific molecular markers, most notably the kit tyrosine kinase receptor and the paired box gene 3 transcription factor (Pax3), but do not express markers associated with melanocyte differentiation such as tyrosinase (Tyr) and tyrosinase-related protein 1 (Tyrp1) [10].
The development of these precursor cells, termed melanoblasts, is regulated by numerous signalling pathways and growth factors, including the WNT/β-catenin, the KIT ligand and its receptor (KITL/KIT), endothelins and the endothelin receptor-B (EDNs/EDNRB), the transforming growth factor beta and its receptor (TGF-β/TGF-βR), the α-Melanocyte-stimulating hormone and its receptor (α-MSH/MC1R), and Notch signalling pathways [10].
The B-cell lymphoma 2 (Bcl-2) antiapoptotic protein has been found to be essential for the survival of migrating melanoblasts and their continued viability during development [11].
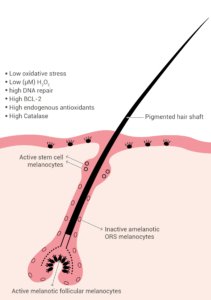
Fully developed hair follicles contain several discrete melanocyte subpopulations distributed in distinct compartments (illustrated in Figure 1) [11]. Poorly differentiated or undifferentiated amelanotic melanoblasts persist in the upper hair follicle reservoir (hair bulge), outer root sheath and proximal hair bulb matrix [11].
Mature melanocytes are present in the infundibulum, the sebaceous gland, and the upper hair bulb matrix around and above the dermal follicular papilla [11].
The mature melanocytes contain melanosomes and have been found to express melogenic enzymes and proteins, including tyrosinase (TYR), TYRP1 and pMEL17 (premelanosome protein 17) [6].
It is the fully differentiated melanocytes in the hair bulb matrix that synthesize and transfer the melanin to precortical keratinocytes for the pigmentation of the growing hair shaft [11].
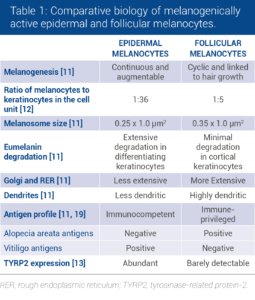
Although follicular and epidermal melanocytes share the same embryonic origin, striking differences in melanocyte cell biology can be observed between the two types of melanocytes (Table 1).
The active melanocytes in the follicular melanin unit are larger, more dendritic, produce larger melanosomes and have more extensive Golgi apparatus and rough endoplasmic reticulum than epidermal melanocytes [11, 12].
Differences in the expression of melanogenic proteins have also been identified: TYRP2 is absent from melanin-producing melanocytes in the hair bulb but is present in the scalp epidermis [13].
Figure 1: Pigmented hair follicle
II. Melanogenesis and the hair growth cycle
The hallmark difference between pigment production in epithelial and follicular melanocytes is that melanogenesis in hair melanocytes is tightly coupled to the hair growth cycle, whereas melanin production in epidermal melanocytes occurs continuously [11, 12].
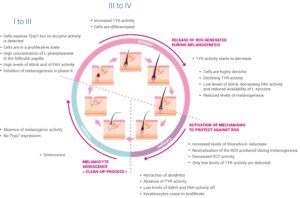
The hair growth cycle consists of three principle phases: the telogen, anagen and catagen phases, with each new cycle requiring the reconstruction of an intact follicular hair unit [10]. The temporal and spatial dynamics of the proliferation and differentiation of the various discrete melanocyte subpopulations during this growth cycle have been tracked by monitoring the expression of key melanocyte markers TYR, TYRP1, cKIT and Ki67 (summarized in Figure 2).
The telogen phase of the cycle is described as the resting stage of hair growth. Although the telogen hair follicle contains all of the cell precursors required for the subsequent anagen growth phase, no melanin is produced during this stage and the cells do not express Tyrp1. Hair growth, active melanogenesis and the transfer of melanin to keratinocytes only occur during the anagen phase of the hair cycle.
Anagen in human scalp hair lasts on average 3 years but can persist for 7 years or more. During the first few days of anagen (anagen I), some of the cell precursors begin to express Tyrp1 and small amounts of protein, although no enzyme activity can be detected. Cells close to the reforming hair bulb enter a state of proliferation. The follicular papilla begins to prepare for melanin synthesis by accumulating high concentrations of L-phenylalanine, a precursor of the L-tyrosine required for melanin synthesis. The level of the allosteric inhibitor of tyrosinase, tetrahydrobiopterin (6BH4), and the activity of the enzyme responsible for the conversion of L-phenylalanine to L-tyrosine, phenylalanine hydroxylase (PAH), also increase during anagen I-II.
Melanogenesis begins towards the end of anagen II and rates of melanin synthesis increase through anagen III, when the levels of 6BH4 fall and TYR activity increases. Hair bulb melanocytes undergo a burst of proliferation during this phase and this proliferation continues until the hair pigmentary unit enters into anagen IV.
Melanocytes in anagen III develop a more extensive Golgi apparatus and a rough endoplasmic reticulum, and become more dendritic in preparation for the melanosome-mediated transfer of melanin to pre-cortical keratinocytes.
Early anagen IV is characterized by maximal hair fiber and melanin production but the rate of melanogenesis begins to slow in late anagen IV, with a notable decline in the availability of L-tyrosine, in the TYR activity and in the expression of other melanogenic proteins as the cycle progresses towards anagen VI. This slow-down may be due to several factors, including changes in the redox status of the hair follicle towards a more reducing environment associated with low levels of 6BH4 and PAH activity and increased activity of thioredoxin reductase during anagen VI. Induction of thioredoxin reductase may allow neutralisation of the reactive oxygen species produced during melanogenesis.
The end of melanogenesis and the beginning of the catagen phase are associated with an absence of TYR activity, and very low levels of 6BH4 and PAH activity, accompanied by the retraction of melanocyte dendrites and shortly afterwards with the end of keratinocyte proliferation in the hair shaft. Extensive apoptosis-driven hair follicle regression occurs during the catagen phase, leading to an absence of melanocytes in the hair bulb. The majority of the melanin-producing melanocytes in the hair bulb do not survive the catagen phase. Although the presence of Pmel17-positive inactive melanocytes in the catagen-associated epithelial column and also in the telogen sac provides evidence for the survival of at least some of the melanocytes that were active in the anagen phase, the current hypothesis is that the immature melanocytes required for the new phase of telogen in the subsequent hair growth cycle are recruited from the melanoblast reservoir in the upper hair follicle, rather than generated from the de-differentiation and re-differentiation of previous generation active melanocytes.
Figure 2: The three principle phases of the hair growth cycle, telogen, anagen and catagen. TYRP1, tyrosinase-related protein-1; 6BH4, tetrahydrobiopterin; PAH, phenylalanine hydroxylase; TYR, tyrosinase; ROS, reactive oxygen species; DCT, dopachrome tautomerase
III. Regulation of melanogenesis and of the hair growth cycle
Control of melanogenesis in hair follicles is complex and involves a regulatory network and signalling pathways that rely on extensive interactions and crosstalk between the subpopulations of cells that make up the hair follicular unit.
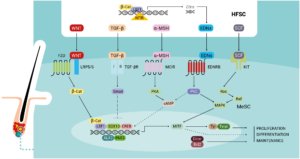
The surrounding hair follicle stem cells provide a functional niche which governs the activation of proliferation, migration and differentiation of melanocyte stem cells during the hair cycle [10]. Melanocyte stem cells express a range of receptors – including Frizzled (FZD), TGF-βR, MC1R, EDNRB and KIT – but the ligands for these receptors – WNT, TGF-β, α-MSH, EDNs and KITL – are expressed in the neighbouring hair follicle stem cells [10]. The WNT/β-catenin signalling pathway is essential for melanocyte stem cell maintenance and differentiation. WNT ligands are expressed in the stem cell niche throughout the telogen phase and the WNT/β-catenin signalling pathway is activated at the start of anagen and suppressed during the remainder of the hair cycle [10].
Activated β-catenin is translocated to the nucleus where it upregulates the melanocyte inducing transcription factor (MITF), a key regulator of genes involved in melanocyte stem cell maintenance (including Bcl-2) and melanogenesis (Tyr and Tyrp1) [10, 14].
KIT signalling is involved in melanocyte development and pigmentation, via the upregulation of MITF [10, 14]. Levels of KIT expression in the hair bulge are low, whereas they are high in hair bulb melanocytes and epithelial cells. KITL is expressed in the dermal papilla, in dermis and in the perifollicular connective tissue sheath [10].
The EDNRB is expressed on melanocyte stem cells, melanocytes and on outer root sheath cells and is involved in stem cell maintenance and proliferation in hair follicles [10]. EDNRB has three ligands: EDN1 and EDN2 are upregulated by β-catenin and expressed during the early anagen phase to regulate melanocyte stem cell proliferation, differentiation and migration, whereas EDN3 is expressed in later anagen and plays a role in promoting melanin synthesis.
The TGF-βR is expressed in the bulge region during late anagen. TGF-β signalling is dependent on Bcl-2, and plays an important role in melanocyte stem cell maintenance by inhibiting the expression of MITF, downregulating genes involved in melanin synthesis and promoting melanocyte stem cell quiescence [10].
MC1R signalling is thought to play a major role in the regulation of follicular melanogenesis [12]. The MC1R is specifically expressed on the surface of melanocytes throughout the anagen phase of the hair cycle [10]. MC1R stimulation in response to α-MSH, derived from pro-opiomelanocortin (POMC) produced locally in keratinocytes, leads to an upregulation of the cAMP pathway and to the activation of numerous downstream signalling pathways, thus leading to an increased activity of melanogenic enzymes [10, 14].
Finally, the NOTCH signalling pathway also appears to play an important role in melanocyte stem cell maintenance in hair follicles [10]. In addition to regulating hair cycle-dependent changes in melanogenesis, this complex regulatory network also mediates the response of the melanocyte stem cells in the bulge to wounding and UVB exposure: the α-MSH/MC1R pathway is activated in response to these stressors which induce melanocyte stem cell differentiation and epidermal migration [10].
Follicular melanogenesis is also influenced by numerous intrinsic factors such as body distribution, racial and gender differences, hormone responsiveness, genetic defects and age-associated changes [11, 12]. The regulation of melanogenesis, and melanocyte stem cells and of their niche environment (Figure 3), in response to these factors could be mediated by a range of signalling pathways which involve the ligands expressed by neighbouring cells of the hair follicular unit and melanocyte receptors (muscarinic, oestrogen receptors α and β, and α1-adrenergic receptors) [14].
Figure 3: Local regulation of survival, quiescence, proliferation, and differentiation of adult melanocyte stem cells in their niche within the hair bulge.
III. PHYSIOPATHOLOGY OF CANITIES
The melanocytes in hair follicles are more sensitive to the influence of age than the melanocytes in the epidermis, with senile canities being the most overt age-rated change [11]. White hairs begin to appear in individual hair follicle units after 10 hair growth cycles. Numerous theories have been proposed for the mechanisms underlying age-related loss of hair pigmentation (summarized in Figure 4).
One hypothesis is that greying is caused by the effects of age on the melanogenic hair bulb melanocytes in the matrix adjacent to the dermal papilla. Aging is associated with reduced numbers of differentiated and functioning hair bulb melanocytes (Figure 4, hypothesis I), decreased tyrosinase activity and the disruption of the normal interaction between active bulb melanocytes and precortical keratinocytes [15]. The precise mechanisms leading to the loss of active melanocytes in the hair bulb have not been fully elucidated but are likely to involve oxidative stress leading to cellular damage, the onset of replicative senescence and melanocyte death by apoptosis [4, 15]. Residual hair bulb melanocytes in greying hair contain fewer and smaller melanosomes, sometimes packaged within autophagolysosomes, and large vacuoles [11, 15, 16]. The recruitment of dendritic cells (including Langerhans cells) in the hair bulb has also been observed [4, 16]. The reduction of tyrosinase activity in aging hair bulb melanocytes may be due to changes in responsiveness to the neuroendocrine factors that regulate melanogenesis (ACTH, α-MSH, and β-endorphin) [16, 17]. Despite reduced tyrosinase activity, degenerating melanocytes still contain significant amounts of melanin. However, this melanin is not transferred to surrounding precortical keratinocytes suggesting that defective interactions between degenerating hair bulb melanocytes and keratinocytes also contribute to greying [4, 15].
Figure 4: Aged non pigmented hair follicle
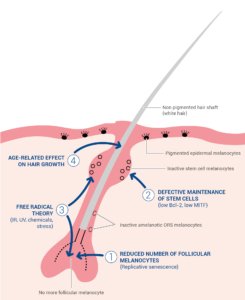
Another hypothesis is that greying results from the defective maintenance of hair follicle stem cells and melanocyte stem cells in the hair bulge (Figure 4, hypothesis II). Failure to maintain the stem cell reservoir appears to be associated with age-related reductions in the antiapoptotic protein Bcl-2, which trigger melanoblast apoptosis as well as an altered activity of the key transcriptional regulator of melanogenesis, MITF, resulting in ectopic differentiation within the hair bulge reservoir [17]. Several of the melanocyte-specific cell markers (SOX10, PAX3, and MITF) are absent in hair bulbs from white hair, suggesting that greying in humans is linked to the depletion of melanoblasts that migrate to the hair bulb [17]. Studies of Col17a1 deficient mice, which display premature greying and hair loss, have highlighted the role played by a reduced niche function in the loss of hair pigmentation [10, 17]. Col17a1 is expressed in follicular stem cells but not in melanocyte stem cells. However, Col17a1 deficiency leads to defective TGF-β signalling from neighbouring keratinocyte stem cells and to premature melanocyte stem cell differentiation thus resulting in the depletion of the stem cell reservoir and premature greying [10, 17].
One of the key factors driving mature melanocyte degeneration in the hair bulb and melanoblast depletion in the hair bulge reservoir is an increasing level of redox imbalance with age, the so-called free-radical theory of greying (Figure 4, hypothesis III) [18]. Environmental (UVR, psychoemotional stress and inflammation) and endogenous (abundant production of H2O2 and other free radicals during melanin synthesis) factors place melanocytes under a high degree of oxidative stress. Oxidative stress leads to increased intracellular levels of reactive oxygen species (ROS) that cause lipid, protein and DNA damage and induce melanocyte apoptosis [18]. When occurring in the context of age-related impairment of antioxidant mechanisms (decreased Bcl-2 expression and catalase activity), the accumulation of ROS appears likely to play a major role in the depletion of active melanocytes and melanoblasts and in the pathophysiology of canities [15, 18].
Numerous clinical differences have been identified between pigmented and nonpigmented hair, with white hair growing more rapidly and being thicker than darker hair. Indeed, white hair is associated with the increased expression of several genes involved in active hair growth (notably keratins and keratin-associated proteins) [17]. These age-related changes in hair growth could result from reduced melanosome activity and decreased melanin transfer to keratinocytes. Melanosomes (through their ability to act as a calcium buffer), melanocytes (as producers of regulatory factors such as cytokines, growth factors etc.) and melanin transfer (leading to a reduction in keratinocyte proliferation) appear to form a regulatory package that influence the behaviour of neighbouring precortical keratinocytes [15]. Age-related increases in active hair growth (Figure 4, hypothesis IV) may be a causative factor in the loss of hair pigmentation and lead to an increased ROS production and to the depletion of active melanocytes and of the melanoblast reservoir [17].
IV. CONCLUSION
In conclusion, despite recent advances in our knowledge of the impact of aging on the hair pigmentary unit, mainly obtained from studies of murine models, further advances in our understanding of melanocyte development, of regulation and of the underlying mechanisms involved in the greying of human hair follicles are required before more effective treatments for senile and premature canities can be developed. In addition to developing new antioxidant therapies and other targeted treatments that could be delivered by the hair follicular route to prevent greying [6], the suppression of hair growth may represent a more effective strategy for the management of canities than attempting to reactivate depleted melanoblast reservoirs [17].
REFERENCES
1. Ito, S. and K. Wakamatsu, Diversity of human hair pigmentation as studied by chemical analysis of eumelanin and pheomelanin.
Journal of the European Academy of Dermatology and Venereology, 2011. 25(12): p. 1369-1380.
2. Rees, J.L., The Melanocortin 1 Receptor (MC1R): More Than Just Red Hair. 2000: Copenhagen.
p. 135-140.
3. Morgan, M.D., et al., Genome-wide study of hair colour in UK Biobank explains most of the SNP heritability. Nature Communications, 2018. 9(1): p. 5271.
4. Tobin, D.J., Aging of the hair follicle pigmentation system. International journal of trichology, 2009. 1(2): p. 83-93.
5. Pandhi, D. and D. Khanna, Premature graying of hair. 2013. 79(5): p. 641-653.
6. Sehrawat, M., et al., Biology of hair pigmentation and its role in premature canities. 2017. 4(1): p. 7-12.
7. Bhat, R.M., et al., Epidemiological and investigative study of premature graying of hair in higher secondary and pre-university school children. International journal of trichology, 2013. 5(1): p. 17-21.
8. Panhard, S., I. Lozano, and G. Loussouarn, Greying of the human hair: a worldwide survey, revisiting the ‘50’ rule of thumb. Br J Dermatol, 2012. 167(4): p. 865-73.
9. Kumar, A., H. Shamim, and U. Nagaraju, Premature graying of hair: Review with updates. 2018. 10(5): p. 198-203.
10. Li, H. and L. Hou, Regulation of melanocyte stem cell behavior by the niche microenvironment. 2018. 31(5): p. 556-569.
11. Tobin, D.J., The cell biology of human hair follicle pigmentation. 2011. 24(1): p. 75-88.
12. Tobin, D.J., Human hair pigmentation–biological aspects. Int J Cosmet Sci, 2008. 30(4): p. 233-57.
13. Commo, S., et al., Absence of TRP-2 in Melanogenic Melanocytes of Human Hair. 2004. 17(5): p. 488-497.
14. D’Mello, S.A.N., et al., Signaling Pathways
in Melanogenesis. International journal of molecular sciences, 2016. 17(7): p. 1144.
15. Tobin, D.J., Age-related hair pigment loss. Curr Probl Dermatol, 2015. 47: p. 128-38.
16. Tobin, D.J. and R. Paus, Graying: gerontobiology of the hair follicle pigmentary unit. Experimental Gerontology, 2001. 36(1): p. 29-54.
17. Jo, S.K., et al., Three Streams for the Mechanism of Hair Graying. Annals of dermatology, 2018. 30(4): p. 397-401.
18. Arck, P.C., et al., Towards a «free radical theory of graying»: melanocyte apoptosis in the aging human hair follicle is an indicator of oxidative stress induced tissue damage. Faseb j, 2006. 20(9):
p. 1567-9.
19. TOBIN, D.J. and J.-C. BYSTRYN, Different Populations of Melanocytes Are Present in Hair Follicles and Epidermis. Pigment Cell Research, 1996. 9(6): p. 304-310.