
The quantity of data generated by a study of the whole transcriptome with Affymetrix microarray can make result analysis and processing complex. Bioalternatives can assist you in all the development phases of the bioinformatics analysis.
Processing of whole transcriptome raw data
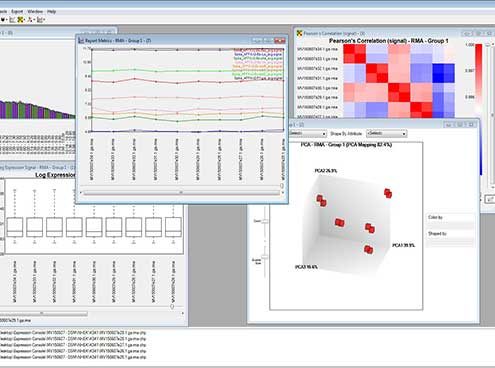
Raw data which correspond to the translation of signal intensity into numerical values are generated as a .CEL file, a specific format of the Affymetrix® platform. Raw data are, at this stage, hosted on a dedicated server and linked to Ba LIMS to ensure their traceability and their security. Raw data are submitted to quality control for labeling and for hybridization and to a first normalization step by using the dedicated Expression Console™ Software.
Standard analysis of data
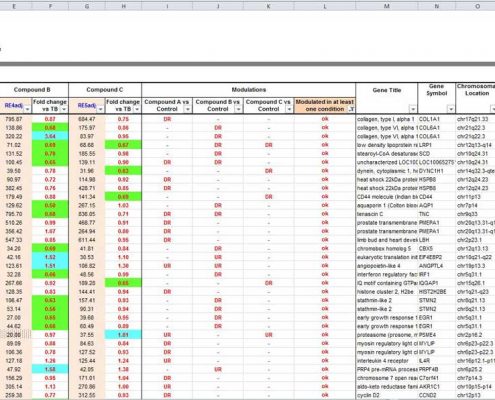
A bioinformatics analysis performed with fold change calculation and statistical tests highlights the significantly modulated genes between different conditions. This first level of data mining is provided with graphical representations generated by the Affymetrix® Transcriptome Analysis Console (TAC) Software:
- ranking of modulated genes (up/down)
- scatter plots (representation of global effects)
and optionally:
- Venn diagram
- statistical analysis (mean, ANOVA or t-test)
Basic functional analysis: biological process
An extensive bioinformatics mining carried out using the D.A.V.I.D. (Database for Annotation, Visualization and Integrated Discovery) database allows the most affected biological processes to be identified.
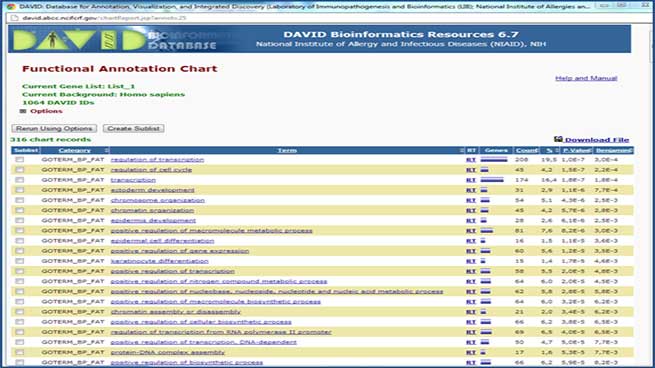
Advanced functional analysis: pathway and clustering
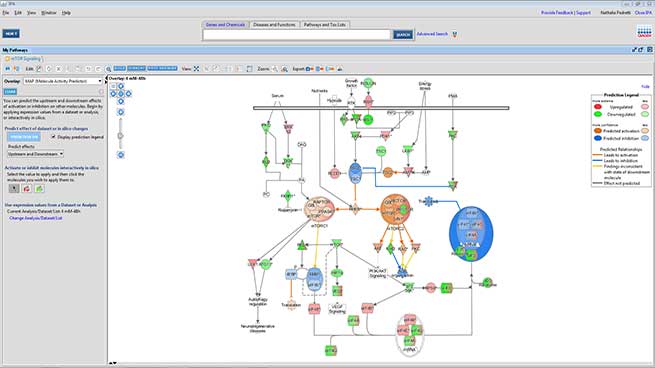
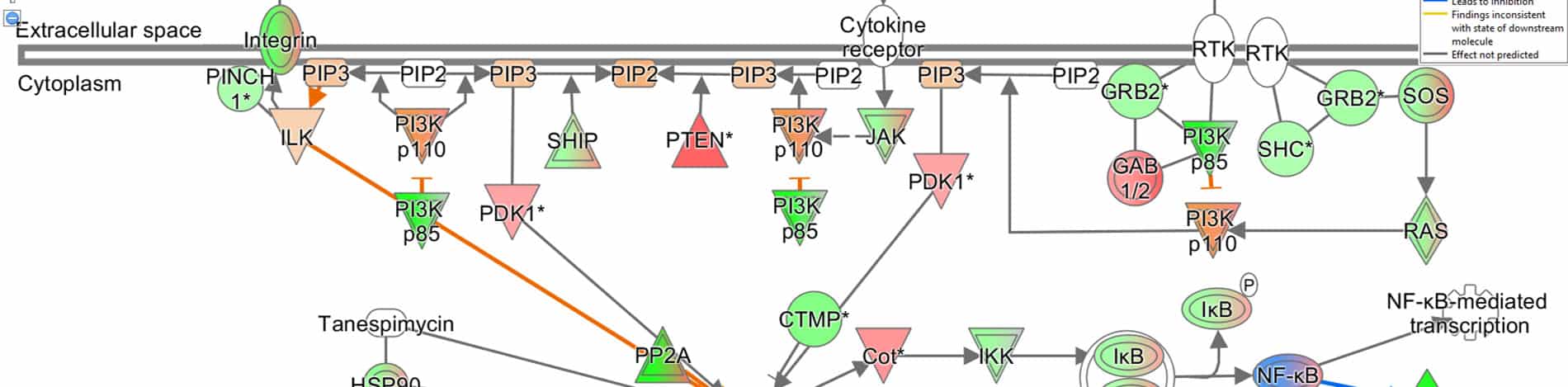
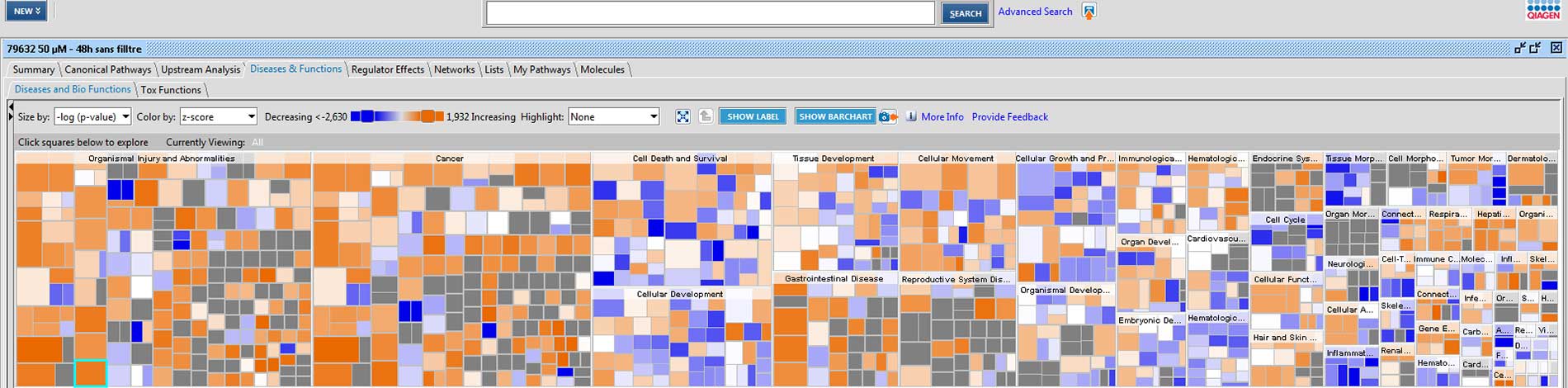
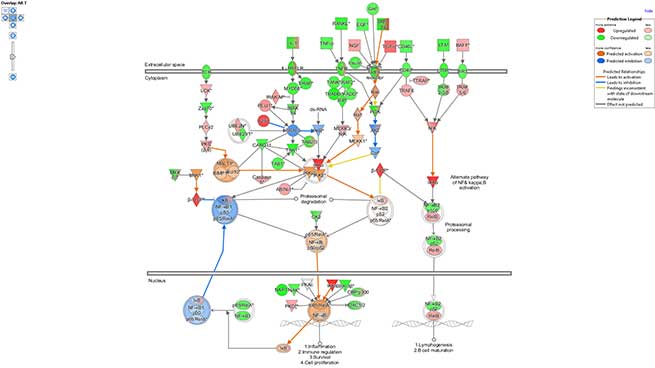
A more advanced bioinformatics analysis performed using the IPA (Ingenuity Pathway Analysis) software by Qiagen, allows the identification of the most affected signaling pathways and, when possible, the prediction of their modulation direction.
{kind=link}