



La majorité des personnes ayant deux variants de MC1R avaient les cheveux blonds ou brun clair. Plusieurs autres locus associés à la couleur des cheveux roux ont été identifiés dans cette étude, y compris des variations du gène codant pour la protéine de signalisation agouti (ASIP), un agoniste inverse de MC1R. Plus de 200 variants génétiques ont également été associés de manière indépendante à un spectre de couleurs de cheveux allant du noir au blond. Un grand nombre de ces gènes sont impliqués dans la croissance et la texture des cheveux plutôt que dans la pigmentation, soulignant le rôle joué par les interactions mélanocytes et kératinocytes dans la couleur des cheveux [3].
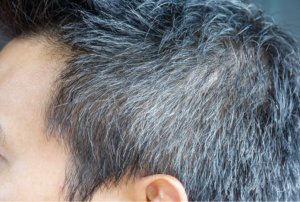
Des changements significatifs de la pigmentation des cheveux se produisent avec l’âge, avec un assombrissement des cheveux au cours de l’enfance, de l’adolescence et du jeune adulte en réponse aux hormones, notamment les stéroïdes sexuels, suivis du vieillissement des cheveux en fonction de l’âge [4]. Ce grisonnement, appelé canitie sénile, est généralement observé pour la première fois au niveau des tempes et du vertex chez les hommes et à la naissance des cheveux chez les femmes, avant de s’étendre sur le reste du cuir chevelu [5]. La canitie est définie comme la perte progressive de pigmentation naturelle des cheveux au fil du temps provoquant un mélange de cheveux foncés normalement pigmentés et de cheveux hypomélanotiques et amélanotiques plus clairs, aboutissant à une dilution de la couleur des cheveux et un blanchissement progressif perçu comme grisonnant [6]. Une canitie prématurée peut également survenir, parfois dès l’adolescence [7].
La canitie sénile survient chez les hommes et les femmes, avec une incidence de 78% et 71% respectivement chez les hommes et les femmes adultes d’âge moyen (45 à 65 ans) [8]. L’incidence des cheveux blancs varie en fonction de l’origine ethnique ou géographique et de la couleur naturelle des cheveux, les incidences les plus faibles étant rapportées dans les populations où prédominent les cheveux noirs [8]. L’âge moyen d’apparition de la canitie sénile dans les différents groupes ethniques va du milieu à la fin de la trentaine respectivement chez les Caucasiens et les Asiatiques, au milieu de la quarantaine en Afrique [4]. Par conséquent, l’âge auquel le grisonnement est défini comme canitie précoce varie également : avant l’âge de 20 ans chez les Caucasiens, 25 ans chez les Asiatiques et 30 ans chez les Africains [4].

II. Causes sous-jacentes de la canitie
Le stress psycho-émotionnel, les maladies associées à une carence ou une malabsorption de protéines, ainsi que diverses carences en vitamines (vitamine D3 et vitamine B12) et en minéraux (calcium, cuivre et fer ou ferritine) sont tous associés à une probabilité accrue de grisonnement prématuré [6, 7]. L’utilisation d’agents topiques (dithranol, chrysarobine et analogues de la prostaglandine F2-alpha) et de médicaments (chloroquine, méphénésine, interféron alpha et imatinib) peut également déclencher un grisonnement prématuré [5]. De plus, des facteurs environnementaux – l’exposition aux rayons ultraviolets (UV), aux pesticides et au tabac – influencent l’apparition de la canitie [6].
Les facteurs génétiques jouent un rôle majeur dans la canitie. Le grisonnement prématuré en l’absence de toute pathologie sous-jacente est commun et est un caractère héréditaire dominant autosomique. En outre, de nombreuses pathologies sont associées à un grisonnement prématuré, notamment des maladies auto-immunes (vitiligo, anémie pernicieuse et troubles de la thyroïde), des syndromes de vieillissement prématuré (progéria, syndrome de Werner et syndrome de Rothmund-Thomson), la diathèse atopique et le syndrome de Down [5, 6, 9 ].
III. Traitements de la canitie
La canitie prématurée est une motivation courante pour demander la consultation d’un dermatologue. La réversibilité de l’hypopigmentation associée à certaines carences nutritionnelles a été rapportée avec un traitement approprié pour l’étiologie sous-jacente, telle que la supplémentation en fer chez les patients anémiques [7]. De nombreux traitements contenant une grande variété de molécules (acide para-aminobenzoïque, acide phytique, capixyle, pantothénate de calcium, etc.) ont été commercialisés pour la canitie, mais les résultats sont souvent contradictoires et seulement temporaires [6]. Le traitement consiste souvent à camoufler les cheveux blancs avec des colorants. Certaines études ont montré que les colorants capillaires protégeaient les cheveux des dégradations liées aux rayons ultraviolets [6, 9]. Des approches thérapeutiques plus prometteuses font appel à des composés topiques anti-vieillissement / anti-oxydants, à l’hormone de croissance recombinante humaine et à l’utilisation de l’acétylhexapeptide-1 (Melitane ™), agoniste de la mélanocortine (a-MSH) [6, 9]. Les thérapies futures reposeront sur l’identification des gènes et des protéines cibles du follicule pileux et pourront également utiliser des liposomes pour administrer de manière sélective des traitements topiques au follicule pileux [6].
II. LA BIOLOGIE DE LA PIGMENTATION DES CHEVEUX
I. Développement des mélanocytes et unité pigmentaire du follicule pileux
Les mélanocytes folliculaires sont dérivés de cellules souches de mélanocytes exprimant des marqueurs moléculaires spécifiques du lignage mélanocytaire, notamment le récepteur de tyrosine kinase kit et le facteur de transcription paired box 3 (Pax3), mais pas les marqueurs associés à la différenciation des mélanocytes telle que la tyrosinase (Tyr) et la protéine 1 liée à la tyrosinase (Tyrp1) [10]. Le développement de ces cellules précurseurs appelées mélanoblastes est régulé par de nombreuses voies de signalisation et facteurs de croissance, notamment le WNT / β-caténine, le ligand KIT et son récepteur (KITL / KIT), les endothélines et le récepteur de l’endothéline-B (EDNs / EDNRB), le facteur de croissance TGF-β et son récepteur (TGF-βR), La mélanocortine et son récepteur (α-MSH / MC1R) et les voies de signalisation de Notch [10]. Enfin, la protéine anti-apoptotique BCL-2 du (lymphome à cellules B) s’avère essentielle pour la survie des mélanoblastes en migration et leur viabilité persistante continue au cours du développement [11].
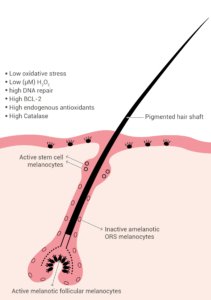
Les follicules pileux complètement développés contiennent plusieurs sous-populations distinctes de mélanocytes réparties dans des compartiments différents (illustrés à la figure 1) [11]. Des mélanoblastes amélanotiques mal différenciés ou indifférenciés persistent dans le réservoir supérieur du follicule pileux (renflement du cheveu), la gaine radiculaire externe et la partie proximale de la matrice du bulbe pileux [11]. Des mélanocytes matures sont présents dans l’infundibulum, les glandes sébacées et la partie supérieure de la matrice du bulbe pileux autour et au-dessus de la papille folliculaire dermique [11]. Les mélanocytes matures contiennent des mélanosomes et se sont révélés exprimer des protéines et des enzymes mélogéniques, notamment TYR, TYRP1 et pMel17 [6]. Ce sont les mélanocytes totalement différenciés de la matrice du bulbe pileux qui synthétisent et transfèrent la mélanine aux kératinocytes précorticaux pour la pigmentation de la tige pilaire en croissance [11].
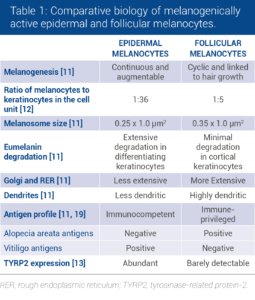
Bien que les mélanocytes folliculaires et épidermiques partagent la même origine embryonnaire, des différences frappantes dans la biologie cellulaire des mélanocytes peuvent être observées entre les deux types de mélanocytes (Tableau 1). Les mélanocytes actifs dans l’unité mélanique folliculaire sont plus gros, plus dendritiques, produisent des mélanosomes plus gros et ont un appareil de Golgi et un réticulum endoplasmique rugueux plus étendus que les mélanocytes épidermiques [11, 12]. Des différences d’expression des protéines mélanogènes ont également été identifiées: TYRP-2 est absent des mélanocytes produisant de la mélanine dans le bulbe pileux, mais est présent dans l’épiderme du cuir chevelu [13].
Figure 1: Follicule pileux pigmenté jeune
II. Mélanogénèse et cycle de croissance des cheveux
La différence principale entre la production de pigment dans les mélanocytes épithéliaux et folliculaires est que la mélanogénèse dans les mélanocytes pileux est étroitement liée au cycle de croissance des cheveux, alors que la production de mélanine dans les mélanocytes épidermiques est continue [11, 12].
Le cycle de croissance des cheveux comprend trois phases principales : télogène, anagène et catagène, chaque nouveau cycle nécessitant la reconstruction d’une unité capillaire folliculaire intacte [10]. La dynamique temporelle et spatiale de la prolifération et de la différenciation des différentes sous-populations discrètes de mélanocytes au cours de ce cycle de croissance a été suivie en surveillant l’expression des marqueurs clés des mélanocytes TYR, TYRP1, cKIT et Ki67 (résumés à la Figure 2).
La phase télogène du cycle est décrite comme étant la phase de repos de la croissance des cheveux. Bien que le follicule pileux télogène contienne tous les précurseurs cellulaires nécessaires à la phase de croissance anagène ultérieure, aucune mélanine n’est produite au cours de cette étape et les cellules n’expriment pas Tyrp1. La croissance des cheveux, la mélanogenèse active et le transfert de mélanine aux kératinocytes ne se produisent que pendant la phase anagène du cycle pilaire. La phase anagène dans la chevelure humaine dure en moyenne 3 ans mais peut persister pendant 7 ans ou plus. Au cours des premiers jours de cette phase (anagène I), certains précurseurs cellulaires commencent à exprimer Tyrp1 et de petites quantités de protéines, bien qu’aucune activité enzymatique ne puisse être détectée. Les cellules proches du bulbe pileux en train de se reformer entrent dans un état de prolifération. La papille folliculaire commence à se préparer à la synthèse de mélanine en accumulant de fortes concentrations de L-phénylalanine, précurseur de la L-tyrosine nécessaire à la synthèse de mélanine. Le taux d’inhibiteur allostérique de la tyrosinase, la tétrahydrobioptérine (6BH4), et l’activité de la phénylalanine hydroxylase (HAP), l’enzyme responsable de la conversion de la L-phénylalanine en L-tyrosine, augmentent également au cours des stades anagène I-II. La mélanogenèse commence vers la fin du stade anagène II et les taux de synthèse de la mélanine augmentent avec l’anagène III, lorsque les niveaux de 6BH4 diminuent et que l’activité de TYR augmente. Les mélanocytes du bulbe pileux subissent une explosion de prolifération au cours de cette phase et cette prolifération se poursuit jusqu’à ce que l’unité pigmentaire du cheveu entre en anagène IV. Les mélanocytes dans l’anagène III développent un appareil de Golgi et un réticulum endoplasmique rugueux plus étendus, et deviennent plus dendritiques en préparation du transfert de mélanine induit par les mélanosomes dans les kératinocytes pré-corticaux. Le début de l’anagène IV est caractérisé par une production maximale de fibres de cheveux et de mélanine, mais le taux de mélanogénèse commence à ralentir à la fin de l’anagène IV, avec une diminution notable de la disponibilité de la L-tyrosine, de l’activité de TYR et de l’expression d’autres protéines mélanogènes à mesure que le cycle progresse vers l’anagène VI. Ce ralentissement peut être dû à plusieurs facteurs, notamment les modifications du statut redox du follicule pileux vers un environnement plus réducteur associé à de faibles niveaux d’activité de 6BH4 et de HAP et à une activité accrue de la thiorédoxine réductase au cours de l’anagène VI. L’induction de la thiorédoxine réductase peut permettre la neutralisation des espèces réactives de l’oxygène produites au cours de la mélanogénèse. La fin de la mélanogénèse et le début de la phase catagène sont associés à une absence d’activité de TYR et à de très faibles niveaux d’activité de 6BH4 et d’HAP, accompagnés d’une rétraction des dendrites mélanocytaires et peu de temps après à la fin de la prolifération des kératinocytes dans la tige du cheveu. Une régression extensive du follicule pileux induite par l’apoptose survient pendant la phase catagène, entraînant une absence de mélanocytes dans le bulbe pileux. La majorité des mélanocytes produisant de la mélanine dans le bulbe pileux ne survivent pas à la phase catagène. Bien que la présence de mélanocytes inactifs positifs à la protéine prémélanosomale 17 (Pmel17 / gp100) dans la colonne épithéliale associée à la phase catagène et également dans le sac télogène fournisse la preuve de la survie d’au moins certains des mélanocytes actifs au cours de la phase anagène, l’hypothèse actuelle est que les mélanocytes immatures nécessaires à la nouvelle phase télogène dans le cycle suivant de croissance du cheveu sont recrutés dans le réservoir de mélanoblastes situé dans le follicule pileux supérieur, plutôt qu’ils proviennent de la dédifférenciation et de la redifférenciation de la génération précédente de mélanocytes actifs.
Figure 2: Les trois phases principales du cycle de croissance des cheveux, télogène, anagène et catagène. TYRP1, protéine 1 liée à la tyrosinase; 6BH4, tétrahydrobioptérine; PAH, phénylalanine hydroxylase; TYR, tyrosinase; ROS, espèces réactives de l’oxygène; DCT, dopachrome tautomérase
III. Régulation de la mélanogénèse et du cycle de croissance des cheveux
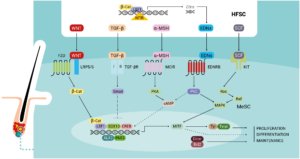
Le contrôle de la mélanogénèse dans les follicules pileux est complexe, impliquant un réseau de régulation et des voies de signalisation qui reposent sur des interactions et des interconnections étendues entre les sous-populations cellulaires qui constituent l’unité folliculaire du cheveu. Les cellules souches du follicule pileux environnantes constituent une niche fonctionnelle qui régit l’activation de la prolifération, la migration et la différenciation des cellules souches mélanocytaires au cours du cycle pilaire [10]. Les cellules souches de mélanocytes expriment une gamme de récepteurs – notamment Frizzled (FZD), TGF-βR, MC1R, EDNRB et KIT – mais les ligands de ces récepteurs – WNT, TGF-β, α-MSH, EDN et KITL – sont exprimés dans les cellules souches de follicules pileux voisins [10]. La voie de signalisation WNT / β-caténine est essentielle pour le maintien et la différenciation des cellules souches mélanocytaires. Les ligands WNT sont exprimés dans la niche des cellules souches tout au long de la phase télogène et la voie de signalisation WNT / β-caténine est activée au début de l’anagène et supprimée pendant le reste du cycle pilaire [10]. La β-caténine activée est transférée au noyau où elle régule positivement le facteur de transcription induisant les mélanocytes (MITF), un régulateur clé des gènes impliqués dans le maintien des cellules souches des mélanocytes (y compris Bcl2) et la mélanogénèse (Tyr et Tyrp1) [10, 14]. La voie de signalisation KIT est impliquée dans le développement et la pigmentation des mélanocytes, via la régulation positive de MITF [10, 14]. Les niveaux d’expression de KIT dans le bulge capillaire sont faibles, alors qu’ils sont élevés dans les mélanocytes du bulbe pileux et les cellules épithéliales. KITL est exprimé dans la papille dermique, le derme et la gaine du tissu conjonctif périfolliculaire [10]. L’EDNRB est exprimé sur les cellules souches de mélanocytes, les mélanocytes et les cellules de l’enveloppe externe de la racine et est impliqué dans le maintien et la prolifération des cellules souches dans les follicules pileux [10]. EDNRB a trois ligands : EDN1 et EDN2 sont régulés positivement par la β-caténine et sont exprimés au début de la phase anagène pour réguler la prolifération, la différenciation et la migration des cellules souches des mélanocytes, alors que EDN3 est exprimé en phase anagène tardive et joue un rôle dans la synthèse de la mélanine. Le TGF-βR est exprimé dans la région du renflement à la fin de l’anagène. La signalisation de TGF-β dépend de BCL2 et joue un rôle important dans le maintien des cellules souches mélanocytaires en inhibant l’expression du MITF, en régulant négativement les gènes impliqués dans la synthèse de la mélanine et en favorisant la mise au repos des cellules souches mélanocytaires [10]. La signalisation MC1R semble jouer un rôle majeur dans la régulation de la mélanogénèse folliculaire [12]. La MC1R est spécifiquement exprimée à la surface des mélanocytes tout au long de la phase anagène du cycle pilaire [10]. La stimulation de MC1R en réponse à l’α-MSH dérivée de la pro-opiomélanocortine (POMC) produite localement dans les kératinocytes, conduit à une régulation positive de la voie de l’AMPc et à l’activation de nombreuses voies de signalisation en aval, entraînant une activité accrue des enzymes mélanogènes [10, 14]. Enfin, la voie de signalisation NOTCH semble également jouer un rôle important dans le maintien des cellules souches mélanocytaires dans les follicules pileux [10].
En plus de réguler les modifications de la mélanogenèse dépendantes du cycle pilaire, ce réseau de régulation complexe intervient également dans la réponse à la blessure et à l’exposition UVB des cellules souches mélanocytaires du bulge : la voie α-MSH / MC1R est activée en réponse à ces facteurs de stress induisant la différenciation et la migration épidermique des cellules souches mélanocytaires [10]. La mélanogenèse folliculaire est également influencée par de nombreux facteurs intrinsèques tels que la distribution corporelle, les différences de race et de sexe, la sensibilité aux hormones, les défauts génétiques et les changements associés à l’âge [11, 12]. La régulation de la mélanogénèse et de l’environnement des cellules souches mélanocytaires et de leur niche (Figure 3) pourrait, en réponse à ces facteurs, être médiée par toute une gamme de voies de signalisation impliquant des ligands exprimés par des cellules voisines de l’unité folliculaire pileuse et des récepteurs des mélanocytes (récepteurs muscariniques et œstrogènes α et β et récepteurs α1-adrénergiques) [14].
Figure 3: Régulation locale de la survie, de la quiescence, de la prolifération et de la différenciation des cellules souches de mélanocytes adultes dans leur niche à l’intérieur du bulge pileux.
III. PHYSIOPATHOLOGIE DE LA CANITIE
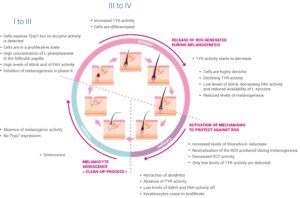
Les mélanocytes des follicules pileux sont plus sensibles à l’influence de l’âge que les mélanocytes de l’épiderme, la canitie sénile étant le changement le plus manifeste avec l’âge [11]. Les cheveux blancs commencent à apparaître dans des unités individuelles du follicule pileux après 10 cycles de croissance des cheveux. De nombreuses théories ont été proposées concernant les mécanismes sous-jacents à la perte de pigmentation des cheveux liée à l’âge (résumés dans la figure 4).
Une hypothèse est que le grisonnement est provoqué par les effets de l’âge sur les mélanocytes mélanogènes du bulbe pileux dans la matrice adjacente à la papille dermique. Le vieillissement est associé à une réduction du nombre de mélanocytes du bulbe pileux différenciés et actifs (Figure 4, hypothèse I), à une diminution de l’activité de la tyrosinase et à la perturbation de l’interaction normale entre les mélanocytes actifs du bulbe et les kératinocytes précorticaux [15]. Les mécanismes précis conduisant à la perte de mélanocytes actifs du bulbe pileux ne sont pas encore complètement élucidés, mais semblent vraisemblablement impliquer un stress oxydatif menant à des lésions cellulaires, l’apparition d’une sénescence réplicative et la mort des mélanocytes par apoptose [4, 15]. Les mélanocytes résiduels du bulbe pileux dans les cheveux grisonnants contiennent des mélanosomes moins nombreux et plus petits, parfois contenus dans des autophagolysosomes, ainsi que de grandes vacuoles [11, 15, 16]. Le recrutement de cellules dendritiques (y compris les cellules de Langerhans) dans le bulbe pileux a également été observé [4, 16]. La réduction de l’activité de la tyrosinase dans les mélanocytes de bulbes pileux vieillissants peut être due à des modifications de la réactivité aux facteurs neuroendocriniens régulant la mélanogénèse (ACTH, α-MSH et β-endorphine) [16, 17]. Malgré une activité réduite de la tyrosinase, les mélanocytes dégénérescents contiennent toujours des quantités importantes de mélanine. Cependant, cette mélanine n’est pas transférée aux kératinocytes pré-corticaux environnants, ce qui suggère que des interactions défectueuses entre les mélanocytes du bulbe pileux dégénératif et les kératinocytes contribuent également au vieillissement. [4, 15].
Figure 4: Follicule pileux âgé non pigmenté
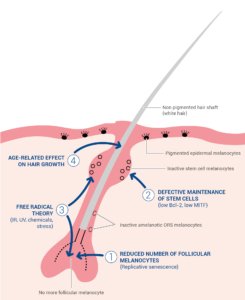
Une autre hypothèse est que le grisonnement résulte d’un mauvais entretien des cellules souches du follicule pileux et des cellules souches mélanocytaires dans le bulge du cheveu (Figure 4, hypothèse II). L’incapacité à maintenir le réservoir de cellules souches semble être associée à une baisse liée à l’âge de la protéine anti-apoptotique BCL2, déclenchant une apoptose des mélanoblastes et une activité modifiée de MITF, le régulateur transcriptionnel clé de la mélanogénèse, ce qui entraîne une différenciation ectopique dans le réservoir du bulge pileux [17]. Plusieurs des marqueurs cellulaires spécifiques aux mélanocytes (SOX10, PAX3 et MITF) sont absents dans les bulbes pileux des cheveux blancs, suggérant que le grisonnement chez l’homme est lié à la diminution des mélanoblastes migrant vers le bulbe pileux [17]. Des études sur des souris déficientes en COL17a1, qui présentent un grisonnement prématuré et une perte de cheveux, ont mis en évidence le rôle joué par la fonction de niche réduite dans la perte de pigmentation des cheveux [10, 17]. Col17A1 est exprimé dans les cellules souches folliculaires mais pas dans les cellules souches de mélanocytes. Cependant, une carence en Col17A1 conduit à une signalisation défectueuse du TGF-β par les cellules souches kératinocytaires voisines et à une différenciation prématurée des cellules souches mélanocytaires, entraînant un épuisement du réservoir de cellules souches et un grisonnement prématuré [10, 17].
L’un des facteurs clés responsables de la dégénérescence mature des mélanocytes dans le bulbe pileux et de l’épuisement en mélanoblastes dans le réservoir du bulbe pileux est un niveau accru de déséquilibre rédox avec l’âge, appelé théorie du grisonnement par les radicaux libres (Figure 4, hypothèse III) [18]. Des facteurs environnementaux (UV, stress psychoémotionnel et inflammation) et endogènes (production abondante de H2O2 et d’autres radicaux libres lors de la synthèse de mélanine) soumettent les mélanocytes à un degré élevé de stress oxydatif. Le stress oxydatif entraîne une augmentation des niveaux intracellulaires d’espèces réactives de l’oxygène (ROS) qui dégradent les lipides, les protéines et l’ADN et provoquent l’apoptose des mélanocytes [18]. Lorsqu’elle survient dans un contexte de dégradation des mécanismes antioxydants liée à l’âge (diminution de l’expression de BCL-2 et de l’activité de la catalase), l’accumulation de ROS semble vraisemblablement jouer un rôle majeur dans l’épuisement en mélanocytes et mélanoblastes actifs et dans la physiopathologie de la canitie [15, 18].
De nombreuses différences cliniques ont été identifiées entre les cheveux pigmentés et non pigmentés, les cheveux blancs se développant plus rapidement et étant plus épais que les cheveux plus foncés. En effet, les cheveux blancs sont associés à une expression accrue de plusieurs gènes impliqués dans la croissance des cheveux (notamment les kératines et les protéines associées aux kératines) [17]. Ces changements liés à l’âge de la croissance des cheveux pourraient résulter d’une activité mélanosomique réduite et d’un transfert diminué de la mélanine aux kératinocytes. Les mélanosomes (grâce à leur capacité à agir en tant que tampon calcique), les mélanocytes (en tant que producteurs de facteurs régulateurs tels que les cytokines, les facteurs de croissance, etc.) et le transfert de mélanine (entraînant une réduction de la prolifération des kératinocytes) semblent constituer un ensemble régulateur qui influence le comportement des kératinocytes précorticaux voisins [15]. L’augmentation avec l’âge de la croissance pileuse active (Figure 4, hypothèse IV) peut être un facteur déterminant de la perte de pigmentation des cheveux, entraînant une production accrue de ROS et un épuisement des mélanocytes actifs et du réservoir de mélanoblastes [17].
IV. CONCLUSION
En conclusion, malgré les progrès récents dans notre connaissance de l’impact du vieillissement sur l’unité pigmentaire du cheveu principalement obtenues à partir d’études de modèles murins, d’autres avancées dans notre compréhension du développement des mélanocytes, de la régulation et des mécanismes sous-jacents impliqués dans le grisonnement des follicules pileux sont nécessaires avant de pouvoir mettre au point des traitements efficaces contre la canitie sénile et prématurée. En plus de développer de nouveaux traitements antioxydants et d’autres traitements ciblés qui pourraient être administrés par la voie folliculaire des cheveux pour prévenir le grisonnement [6], la suppression de la croissance des cheveux peut représenter une stratégie plus efficace pour la prise en charge de la canitie que de tenter de réactiver des réservoirs de mélanoblastes épuisés [17].
RÉFERENCES
1. Ito, S. and K. Wakamatsu, Diversity of human hair pigmentation as studied by chemical analysis of eumelanin and pheomelanin.
Journal of the European Academy of Dermatology and Venereology, 2011. 25(12): p. 1369-1380.
2. Rees, J.L., The Melanocortin 1 Receptor (MC1R): More Than Just Red Hair. 2000: Copenhagen.
p. 135-140.
3. Morgan, M.D., et al., Genome-wide study of hair colour in UK Biobank explains most of the SNP heritability. Nature Communications, 2018. 9(1): p. 5271.
4. Tobin, D.J., Aging of the hair follicle pigmentation system. International journal of trichology, 2009. 1(2): p. 83-93.
5. Pandhi, D. and D. Khanna, Premature graying of hair. 2013. 79(5): p. 641-653.
6. Sehrawat, M., et al., Biology of hair pigmentation and its role in premature canities. 2017. 4(1): p. 7-12.
7. Bhat, R.M., et al., Epidemiological and investigative study of premature graying of hair in higher secondary and pre-university school children. International journal of trichology, 2013. 5(1): p. 17-21.
8. Panhard, S., I. Lozano, and G. Loussouarn, Greying of the human hair: a worldwide survey, revisiting the ‘50’ rule of thumb. Br J Dermatol, 2012. 167(4): p. 865-73.
9. Kumar, A., H. Shamim, and U. Nagaraju, Premature graying of hair: Review with updates. 2018. 10(5): p. 198-203.
10. Li, H. and L. Hou, Regulation of melanocyte stem cell behavior by the niche microenvironment. 2018. 31(5): p. 556-569.
11. Tobin, D.J., The cell biology of human hair follicle pigmentation. 2011. 24(1): p. 75-88.
12. Tobin, D.J., Human hair pigmentation–biological aspects. Int J Cosmet Sci, 2008. 30(4): p. 233-57.
13. Commo, S., et al., Absence of TRP-2 in Melanogenic Melanocytes of Human Hair. 2004. 17(5): p. 488-497.
14. D’Mello, S.A.N., et al., Signaling Pathways
in Melanogenesis. International journal of molecular sciences, 2016. 17(7): p. 1144.
15. Tobin, D.J., Age-related hair pigment loss. Curr Probl Dermatol, 2015. 47: p. 128-38.
16. Tobin, D.J. and R. Paus, Graying: gerontobiology of the hair follicle pigmentary unit. Experimental Gerontology, 2001. 36(1): p. 29-54.
17. Jo, S.K., et al., Three Streams for the Mechanism of Hair Graying. Annals of dermatology, 2018. 30(4): p. 397-401.
18. Arck, P.C., et al., Towards a «free radical theory of graying»: melanocyte apoptosis in the aging human hair follicle is an indicator of oxidative stress induced tissue damage. Faseb j, 2006. 20(9):
p. 1567-9.
19. TOBIN, D.J. and J.-C. BYSTRYN, Different Populations of Melanocytes Are Present in Hair Follicles and Epidermis. Pigment Cell Research, 1996. 9(6): p. 304-310.
